Iš anksto dėkoju!
Taip pat nepamirškite padėkoti savo gydytojams.
genetikas7 22:07
reikia žinoti dėl ko buvo atlikta ekspertizė, kas jam vadovavo ir pamatyti išvadą.
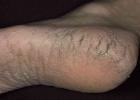
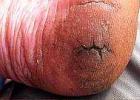
Apžiūros priežastis buvo mano būklė, kai atvykau į kliniką. Man staiga atsirado silpnumas ir praradau kalbą. Kazanėje praėjau visus įmanomus testus ir egzaminus. Nustatyta: progresuojanti leukoencefalopatija, tikriausiai sukelta izoliuoto smegenų vaskulito, pasireiškianti vidutinio sunkumo pažinimo sutrikimais, bulbariniu sindromu, piramidės nepakankamumu. Hiperhomocisteinemija. Hipercholesterolemija. Profesorius rekomendavo atlikti molekulinę genetinę Notch-3 geno mutacijos diagnozę.
Molekulinės genetinės laboratorijos išvadą jau išsiunčiau ankstesniame laiške.
Daktare, padėk man, prašau! Iššifruokite šią išvadą.
genetikas0 20:31
Analizė patvirtino sindromą, kurį įtarė gydytojas.
Labai ačiū už atsakymą. Dabar aš žinau, kad sergu. Kol liga mane visiškai neužvaldė. Matyt, tai ateis vėliau. Na, toks mano likimas.
Vis tiek norėčiau sužinoti, kas yra heterozigotinė mutacija. Akivaizdu, kad tai kažkaip atsispindi ligos paveldėjimo principe. Turiu du vaikus, berniukus. Mano sesuo turi dvi mergaites. Ji už mane jaunesnė, jai 38 metai. Man 44 metai. Liga paveldėjau iš savo tėvo. Jis mirė sulaukęs 61 metų. Mirties priežastis buvo insultas. Jo jaunesnis brolis ir vyresnioji sesuo gyvi ir palyginti sveiki. Jų vaikai taip pat sveiki. Ar tikrai aš vienintelis turiu mutaciją?
Jei atsakysite bent į kelis iš šių klausimų, būsiu jums labai dėkingas.
Viskas kas geriausia.
genetikas3 10:35
Ta pati galimybė egzistavo ir tau, ir tavo seseriai. Kadangi ji yra jaunesnė už jus, kol kas nežinoma, ar ji tai paveldėjo.
Jūsų sesuo ir jūsų vaikai gali atlikti tą patį genetinį tyrimą, kuris buvo atliktas jums. Jei jie nori dabar išsiaiškinti, ar jie paveldėjo mutaciją, ar ne.
heterozigotinė mutacija, kas tai yra
Mthfr
Sveikinimo skiltyje į klausimą: ar galima pagimdyti sveiką vaiką, jei mama turi MTHFR geno mutaciją? paklausė autorė Lyubov Zamkovenko, geriausias atsakymas yra motinos mutacija MTHFR gene – NE VERDIKTAS.
Beje, įvairiose vietose gali būti mutacijų.
Kai mutantinis MTHFR genas aptinkamas heterozigotinėje būsenoje*, nėra jokios įtikinamos priežasties baimintis. Norint išvengti hiperkoaguliacijos būklių, nėštumo metu rekomenduojama kasdien gerti po 0,4 mg folio rūgšties per dvi dozes, gerai maitintis ir kartą per tris mėnesius (arba pagal indikacijas) tirti hemostazogramą.
Dažniausias fermento defektas, susijęs su vidutiniu HC (homocisteino) koncentracijos padidėjimu, yra geno, koduojančio MTHFR, mutacija. MTHFR katalizuoja folio rūgšties pavertimą aktyvia forma. Iki šiol buvo aprašytos 9 MTHFR geno, esančio 1p36.3 lokuse, mutacijos. Dažniausias iš jų yra C677T pakeitimas (MTHFR baltyme – valino pakeitimas alaninu), pasireiškiantis termolabilumu ir MTHFR fermento aktyvumo sumažėjimu. Pastebėta, kad padidinus folatų kiekį maiste, galima išvengti GC koncentracijos padidėjimo plazmoje.
Homocisteino kiekio padidėjimas kraujo plazmoje tiesiogiai koreliuoja su trombomodulino sintezės slopinimu, AT-III ir endogeninio heparino aktyvumo sumažėjimu, taip pat su tromboksano A2 gamybos aktyvavimu. Ateityje tokie pokyčiai sukelia mikrotrombozę ir mikrocirkuliacijos sutrikimus, o tai savo ruožtu vaidina svarbų vaidmenį spiralinių arterijų patologijoje ir akušerinių komplikacijų, susijusių su gimdos placentos kraujotakos pokyčiais, vystymuisi. nuoroda
Padidėjusio homocisteino kiekio kraujyje priežastis: MTHFR geno C677T variantas - fermento metilentetrahidrofolato reduktazės geno mutacija.
Citoziną pakeitus timinu 677 padėtyje, fermento funkcinis aktyvumas sumažėja iki 35% vidutinės vertės.
Polimorfizmo duomenys:
*homozigotų atsiradimo dažnis populiacijoje yra 10-12 proc.
*heterozigotų atsiradimo populiacijoje dažnis – 40 proc.
T varianto nešiotojai nėštumo metu patiria folio rūgšties trūkumą, o tai lemia vaisiaus nervinio vamzdelio vystymosi defektus.
Rūkymas padidina 677T varianto poveikį.
Folio rūgšties vartojimas gali žymiai sumažinti šio polimorfizmo pasekmių riziką.
Apskritai, kas kur bus nuvežtas... Tiksliai pasakyti neįmanoma. Nuo tėvo taip pat priklauso, kas yra jo genome.
Pabandykite užduoti savo klausimą išsamiau čia - nuoroda
Viskas yra Dievo galioje. Čia statistika bejėgė.
heterozigotinė mutacija
Universalus rusų-anglų žodynas. Akademik.ru. 2011 m.
Pažiūrėkite, kas yra „heterozigotinė mutacija“ kituose žodynuose:
genų sintezės mašina – * genus sintezuojanti mašina * genų mašina yra automatinis DNR sintezatorius, skirtas trumpoms (dažniausiai bp ilgio) DNR grandinėms gaminti, naudojamas polimerazės grandininei reakcijai. Genai yra sudėtingi * genai yra sulankstyti * sudėtiniai genai genai ... Genetika. enciklopedinis žodynas
heterodupleksinė analizė d hibridizacija a - heterodupleksinė analizė d hibridizacija a. * heteradupleksinė analizė, p. gibridyzatsyina a. * heterodupleksinė analizė arba h. hibridizacija a. genų mutacijų nustatymo metodas sumaišant mutantinę DNR molekulę, amplifikuotą PGR būdu su DNR... ...Genetika. enciklopedinis žodynas
AUTOSOMINIŲ CHARAKTERISTIKŲ PAVELDĖJIMAS – Laikykime tokią savybę kaip kraujo grupė. Yra keletas kraujo grupių tipų arba sistemų. Labiausiai žinoma AB0 sistema, kuri išskiria keturias pagrindines grupes: I, II, III ir IV; šios grupės taip pat žymimos 0, A, B ir AB, nes ... ... Collier's Encyclopedia
PELĖS – PELĖS yra smulkūs graužikai, kurie kartu su žiurkėmis sudaro Murinae pošeimį. Mus musculus, naminė pelė, yra kosmopolitinė rūšis, išplitusi kartu su žmonėmis visame pasaulyje. Gyvena namuose ir tarnybose; gali gyventi soduose ir krūmuose,... ...Didžioji medicinos enciklopedija
PAVELDIMUMAS – materialių veiksnių, lemiančių organizmo savybių vystymąsi konkrečiomis aplinkos sąlygomis, perdavimo palikuonims reiškinys. N. tyrimo užduotis yra nustatyti atsiradimo, savybių, perdavimo ir... ... Didžiosios medicinos enciklopedijos modelius.
DUMPS arba uridino monofosfato sintetazės trūkumas yra liga, kurią sukelia recesyvinio geno mutacija, atsirandanti dėl „superveislių“ veisimo. c. X. gyvūnaiLiga atsirado JAV ir išplito visame pasaulyje dėl Holšteino veislės galvijų veislinių gyvūnų, embrionų ir spermos pardavimo... ...Genetika. enciklopedinis žodynas
Mes naudojame slapukus, kad suteiktume jums geriausią patirtį mūsų svetainėje. Toliau naudodamiesi šia svetaine sutinkate su tuo. gerai
Mutacijos
F5: Factor V Leiden (G1691A; Arg506Gln)
Aš netyriau kitų mutacijų, kurias turite, jos atrodo kaip folio ciklo mutacijos. Aš išbandžiau kitas mutacijas.
Turite išsitirti homocisteiną.
Tiesioginės temos forume
Kodėl buvo nustatyti silpni embrionai?
Jau valgau ananasus, pirmoje fazėje, po ovuliacijos tęsiu, po truputį. Mielasis labai malonu.
Gladis, Kristinka rašė, kad prieš įlipant jai buvo išrašytas Intralipid. Kiek pamenu.
Populiarūs tinklaraščio įrašai
Situacija tokia, testas teigiamas, dinamika mano nuomone nelabai gera, vakar buvau echoskopu paziureti geltona.
Istorija tokia: siandien 11 delsimo diena, tyrimai dryzuoti, su dinamika, kraujo daviau kovo 5 d., 3870 hCG.
Šiandien yra 12 dpo, matai ką?Tikrinti Mamos čekį arba panelę, trumpai tariant, pigiausias
laikotarpis lygiai 12 savaičių. Ultragarso specialistas visiškai nieko nesakė ((((nors gumbas matosi.
Geriausi straipsniai bibliotekoje
Svetainės medžiagos atkūrimas galimas tik naudojant aktyvią tiesioginę nuorodą į www.babyplan.ru
©17, BabyPlan®. Visos teisės saugomos.
Kas yra heterozigotinė mutacija?
Man 41 metai, man antras nėštumas, pirmas 2010 m. ir baigėsi sveiko vaiko gimimu 2011 m., abu nėštumai buvo IVF (išimti vamzdeliai).
Pirmojo nėštumo metu buvo atlikta krešėjimo faktorių analizė (tiesiog prie kitų tyrimų), nustatytos 3 faktorių mutacijos (Leiden ir dar dviejuose), visos mutacijos heterozigotinės. Gydytojai ne kartą bandė surinkti iš manęs anamnezę – paaiškėjo, kad neturiu jokių krešėjimo problemų apraiškų. Clexane 0,2 per dieną buvo naudojamas maždaug mėnesį po embriono perkėlimo ir daugiau nebuvo naudojamas. Per pirmąjį nėštumą be galo matavosi d-dimeras ir koagulogramos, nėščiosioms viskas buvo normose. Kilo klausimas, ar gimdymą lydėti fraksiparinu, gydytojų nuomonės išsiskyrė, nusprendžiau pasitikėti tais, kurie nesutiko su fraksiparino skyrimu (priežastis ta, kad dėl žemos placentos buvo galimas kraujavimas), gimdymas prasidėjo kaip natūralus, bet buvo atlikta skubi cezario pjūvio operacija. Viskas pavyko gerai. Vienintelė prevencinė priemonė buvo keletą dienų po gimdymo dėvėti specialias kojines. Trombozė ir venos nepasirodė.
Prieš antrąjį IVF kreipiausi į hematologą (rinkausi pagal IVF klinikos rekomendacijas ir pacientų atsiliepimus; apskritai mūsų mieste, kaip suprantu, yra tik 3 hematologai su panašiomis problemomis, o gydytoja pas kurią kreipiausi laikomas gana autoritetingu). Hematologė pasakė, kad man labai didelė komplikacijų rizika, bet kokiu atveju VISĄ nėštumą turėsiu gerti vieną iš fraksiparinų, tik reikia pakoreguoti dozę. Tai, kad su pirmuoju nėštumu viskas gerai, jo nuomone, nieko nereiškia. Ir jis pasakė, kad mums reikia dažniau matuoti d-dimerį. Tačiau jo rekomendacijos skiriasi nuo reprodukcijos specialisto rekomendacijų, nes... Hematologas liepė gerti aspiriną-kardio ir fraksipariną visą pasiruošimo transplantacijai ciklą, bet reprodukcijos specialistė liepė to nedaryti (Clexane buvo paskirta tik po transplantacijos). Todėl tiesiog nežinau, kuo tikėti ir kur ieškoti „tiesos“...
Perskaičiau DUK forume.
1. Vis dėlto, ar būtina profilaktiškai vartoti Clexane (ar kitą fraksipariną) viso nėštumo metu? O gal užtenka būti saugioje pusėje tik pirmąjį trimestrą?
2. Kokia turėtų būti mano taktika (ką ir kaip dažnai tikrinti, į ką atkreipti dėmesį), kad nepraleisčiau, kai tikrai reikia pagalbos?
3. Ir dar, man kyla abejonių, ar išvis yra mutacija... nes man buvo traumų ir, nors ir smulkių, pilvo operacijų (kiaušidės cistos šalinimas, cezario pjūvis), taip pat begalė laparoskopinių operacijų (visokių). kiaušintakių praeinamumo atstatymas, po to vamzdžių pašalinimas) – ar tikrai gali būti, kad niekada nieko nebus? Koks yra efektyviausias mutacijų laboratorinės diagnostikos metodas? (mūsų laboratorijos, kaip suprantu, dirba skirtingais metodais) - Galvoju apie pakartotinį patikrinimą.
Leideno genotipas (krešėjimo faktorius) – heterozigotas
Metilentetrahidrofolato redukto mutacija. - heterozigotas
Protrombino mutacija (krešėjimo faktorius) – heterozigota
Laboratorija, kuri parodė heterozigotines mutacijas, mūsų šalyje laikoma normalia ir patikima, su ja ligoninės bendradarbiauja.
Pasidariau testą dar kartą ir lauksiu rezultato.
(nors abejoju, ar ji testą perlaikė).
"19.13 Trombocitų faktoriai
GP1BA Trombocitų glikoproteinas 1B T-5C, rs) TT Pyromark24
GP1BA trombocitų glikoproteinas 1B (T145M, C>T, rs6065) CC Pyromark24
ITGB3 trombocitų fibrinogeno receptorius (L33P, A1/A2, rs5918) TT Pyromark24
JAK2 Janus kinazė 2 (V617F, G>T, rs) GG Pyromark24
SELPLG P-selektino glikoproteino ligandas (M62I,G>A, rs) GG Pyromark24
Pastaba dėl rezultato: normos variantas.
Pagal šią analizę (2013 m. lapkričio 21 d.) buvo nustatyta:
"19.11 Plazmos krešėjimo faktoriai
plazminogenas (-675 5G>4G,rs) 4G\4G Pyromark24
(iš viso: trys skirtingos analizės su skirtingais rezultatais..)
Įrašo komentarai:
Norėčiau pasikonsultuoti kitu su nėštumu ir krešėjimu susijusiu klausimu.
Apie savo krešėjimo problemas rašiau aukščiau. Nieko naujo blogo nepasirodė, tyrimai normalūs, vaisius ir placenta pagal echoskopiją normalūs, dabar laikotarpis tarp 19 ir 20 savaičių. Turiu nedidelę geležies stokos anemiją (hemoglobinas 103), planuoju profilaktine doze gerti truputį aktiferino. Nustojau naudoti Fraxiparine net po to, kai uždaviau aukščiau pateiktą klausimą šioje temoje. Savo sveikatos būklės pokyčių nepastebėjau.
Kur galiu kreiptis su savo liga?
Imunologijos ir reprodukcijos centras
CIR yra sveikatos teritorija!
Nauja genetika (genomika) nėštumo komplikacijų prevencijai
Daugelio variantų genų ypatumas yra tas, kad jie ilgą laiką gali niekaip nepasireikšti. Patologiniai simptomai gali atsirasti esant papildomoms sąlygoms (mitybos įpročiai, nėštumas, vaistai, gyvenimo būdas ir kt.). Šių papildomų sąlygų išaiškinimas padeda efektyviai užkirsti kelią ligoms ir jų komplikacijoms genetiškai modifikuotų genų nešiotojams.
Trombofilija kaip nėštumo komplikacijų rizikos veiksnys
Trombofilija yra polinkis susidaryti kraujo krešuliams (kraujo krešuliams). Trombofilija gali būti pavojinga gyvybei, jei krešulys blokuoja kraujotaką. Trombofilija gali būti paveldimas sutrikimas, bet gali būti susijęs ir su išorinėmis priežastimis, tokiomis kaip chirurgija, nutukimas, nėštumas, hormoninių kontraceptikų vartojimas, antifosfolipidinis sindromas, padidėjęs homocisteino kiekis ar ilgas nejudrumas. Gydytojai įtaria trombofilija tiems pacientams, kurie anksčiau sirgo tromboze arba kurių artimieji jauname amžiuje (iki 40-50 metų) sirgo tromboze, insultu ar infarktu. Tačiau daugelis žmonių, sergančių trombofilija, neturi jokių simptomų arba simptomai lieka nepastebėti, nes polinkis į trombofiliją nėra pakankamai stiprus. Pastarųjų metų tyrimai parodė, kad trombofilija yra susijusi su padidėjusia nėštumo komplikacijų rizika (pasikartojantis persileidimas, placentos nepakankamumas, vaisiaus augimo sulėtėjimas, vėlyvoji toksikozė (preeklampsija)). Paveldimų trombofilijų genų žymenys apima metilentetrahidrofolato reduktazės mutaciją, Leideno mutaciją ir protrombino geno mutaciją G20210A.
Pastarųjų metų tyrimai parodė, kad persileidimą patyrę pacientai dažnai turi vieną ar daugiau genetinių trombofilijos žymenų. Pavyzdžiui, viename tyrime Leideno mutacija buvo nustatyta 19% persileidusių pacientų, o kontrolinėje grupėje Leideno mutacija buvo nustatyta tik 4% moterų.
Metileno tetrahidrofolato reduktazės mutacija
fermentas su homocisteino metabolizmo sutrikimais. Maždaug tais pačiais metais buvo įrodyta, kad padidėjęs homocisteino kiekis yra nepriklausomas kraujagyslių komplikacijų vystymosi rizikos veiksnys. Pradėtas pastangos išsiaiškinti MTHFR trūkumo genetinį pobūdį. MTHFR geno klonavimas 1993 m. suteikė pagrindą nustatyti mutacijas, susijusias su įvairaus laipsnio šio fermento trūkumu.
Folio ciklas
Fermentas 5,10-metilentetrahidrofolato reduktazė priklauso flavoproteinų grupei ir susideda iš dviejų identiškų subvienetų, kurių molekulinė masė yra apie 70 kDa. MTHFR yra pagrindinis folio ciklo fermentas. Folatas ir folio rūgštis (sintetinis vitaminas, kurio nėra natūraliuose maisto produktuose) yra dvi pteroilglutamo rūgšties (PteGlu) medžiagų šeimos formos. Ši rūgštis yra sudėtinga molekulė, susidedanti iš pteroidinės rūgšties ir vienos (monoglutamatų) arba kelių (iki 9, poliglutamatų) glutamo rūgšties liekanų (žr. 1 pav.). Maisto produktuose, ypač šviežiuose žalumynuose, kepenyse, mielėse ir kai kuriuose vaisiuose, daugiausia yra redukuotų poliglutamatų, kuriuos fermentas pteroilpoliglutamato hidrolazė turi hidrolizuoti iki monoglutamato, kad jie galėtų būti absorbuojami proksimalinėje plonojoje žarnoje. Absorbuotas folio monoglutamatas greitai redukuojamas į tetrahidrofolatą, nes tik redukuotos folio rūgšties formos yra biologiškai aktyvios. Po metilinimo folatas patenka į kraują 5-metiltetrahidrofolato pavidalu. Be maisto, nuolatinį 5-metiltetrahidrofolato tiekimą užtikrina enterohepatinis ciklas: pterilo monoglutamatas absorbuojamas iš žarnyno ir patenka į kepenis, kur redukuojamas ir metilinamas iki 5-metiltetrahidrofolato. Gautas 5-metiltetrahidrofolatas išsiskiria su tulžimi į žarnyną, kur vėliau absorbuojamas ir paskirstomas kraujyje visame kūne.
Į audinį 5-metiltetrahidrofolatas patenka į ląstelę endocitozės būdu, dalyvaujant specifiniams folio receptoriams. Aprašytos trys folio receptorių izoformos. Ląstelės viduje 5-metiltetrahidrofolatas yra metilo grupių donoras ir pagrindinis tetrahidrofolato šaltinis. Pastarasis veikia kaip daugelio monokarboninių grupių akceptorius, virsdamas įvairių tipų folatais, kurie savo ruožtu yra specifiniai kofermentai daugelyje tarpląstelinių reakcijų. Tai 5-formiltetrahidrofolatas (folino rūgštis, leukovorinas), 10-formiltetrahidrofolatas ir 5,10-metilentetrahidrofolatas.
Viena iš reakcijų, kurioms reikalingas 5,10-metilentetrahidrofolatas ir 5-metiltetrahidrofolatas, yra metionino sintezė iš homocisteino (remetilinimo kelias homocisteino metabolizme). Šioje reakcijoje MTHFR atlieka pagrindinį vaidmenį redukuodamas 5,10-metilentetrahidrofolatą iki 5-metiltetrahidrofolato, taip katalizuojantis vienintelę tarpląstelinę reakciją, susidarant 5-metiltetrahidrofolatui. Nors serume ir kituose audinių skysčiuose randama įvairių folatų formų, pagrindinė folio forma plazmoje yra 5-metiltetrahidrofolatas, turintis metilo grupę, reikalingą homocisteinui paversti metioninu. Šioje reakcijoje metilo grupė pirmiausia perkeliama į cob(I)alaminą (vitamino B 12 forma), paverčiant jį metilkobalaminu, kuris vėliau paduoda metilo grupę homocisteinui, sudarydamas metioniną fermento metionino sintaze. Tačiau kai kuriais atvejais cob(I)alaminas gali būti oksiduojamas į cob(II)alaminą, todėl slopinama metionino sintazė. Norint išlaikyti fermentų aktyvumą, būtinas redukcinis metilinimas naudojant fermentą metionino sintazės reduktazę.
Kadangi kobalaminas (vitaminas B 12) yra 5-metiltetrahidrofolato metilo grupės akceptorius, šio vitamino trūkumas sukelia „folio spąstus“. Tai yra medžiagų apykaitos aklavietė, nes metiltetrahidrofolatas negali būti redukuojamas į tetrahidrofolatą ir grąžinamas į folatų telkinį. Nesugebėjimas atkurti metionino sukelia metionino išeikvojimą ir homocisteino pertekliaus išsiskyrimą į kraują.
MTHFR genas
MTHFR genas žmonėms yra ant trumposios 1 chromosomos rankos (1p36.3). Viso kodavimo srities ilgis yra apie 1980 bp. kurio apskaičiuotoji produkto molekulinė masė yra 74,6 kDa. Aminorūgščių seka yra evoliuciškai konservuota, nes yra 90% homologijos su pelių MTHFR polipeptidu. Taip pat buvo iššifruota genominė genomo organizacija. Jį sudaro 11 egzonų, kurių ilgis svyruoja nuo 102 iki 432 bp. ir intronai, kurių ilgis svyruoja nuo 250 iki 1500 bp, išskyrus vieną introną, kurio ilgis 4200 bp.
MTHFR geno polimorfizmas
Aprašyti du MTHFR geno variantai. Labiausiai ištirtas variantas, kuriame nukleotidas citozinas (C) 677 padėtyje, priklausantis 4-jam egzonui, yra pakeistas timidinu (T), dėl kurio folio rūgšties aminorūgšties liekana alaninas pakeičiama valino liekana. surišimo vieta. Šis MTHR polimorfizmas vadinamas C677T mutacija. Asmenys, homozigotiniai šiai mutacijai, pasižymi MTHFR termolabumu ir fermentų aktyvumo sumažėjimu iki maždaug 35% vidutinės vertės. Be to, šiai mutacijai homozigotiniams asmenims yra sutrikęs folatų pasiskirstymas eritrocituose, išreikštas tetraglutamato formilo poliglutamatų ir metilinto tetrahidrofolato darinių kaupimu. Šios mutacijos buvimą lydi padidėjęs homocisteino kiekis kraujyje.
Kitas MTHFR geno polimorfizmo variantas yra nukleotido adenino (A) pakeitimas citozinu (C) 1298 padėtyje. Dėl to fermento reguliavimo srityje glutamino liekana pakeičiama alanino liekana, kurią lydi. šiek tiek sumažėjus aktyvumui. Asmenų, homozigotinių A1298C mutacijos atžvilgiu, MTHFR aktyvumas sumažėja iki maždaug 60 % normalaus. Daroma prielaida, kad fermento aktyvumas sumažėjo dėl fermento reguliavimo pokyčių, kuriuos sukelia jo inhibitorius S-adenozilmetioninas.
Priešingai nei C677T polimorfizmas, A1298C mutacijos heterozigotiškumas ir homozigotiškumas nėra lydimas nei bendro homocisteino koncentracijos padidėjimo, nei folio rūgšties kiekio plazmoje sumažėjimo. Tačiau 677T ir 1298C alelių heterozigotiškumo derinį lydi ne tik fermentų aktyvumo sumažėjimas, bet ir homocisteino koncentracijos padidėjimas plazmoje bei folio koncentracijos sumažėjimas, kaip ir homozigotiškumo 677T atveju.
Alelių 677T ir 1298C homo- ir heterozigotiškumo diagnostika atliekama polimerazės grandininės reakcijos (PGR) metodu.
677T alelio paplitimas
677T alelis yra plačiai paplitęs populiacijoje. Europos rasėje homozigotiškumo dažnis yra apie 10-12%, heterozigotiškumas – apie 40%. Yra didelių tarprasinių ir etninių skirtumų. Dažniausiai šis genas aptinkamas europiečiams, rečiausiai – juodiesiems afrikiečiams, Australijos ir Šri Lankos aborigenams.
Europoje mažiausias 677T alelio dažnis yra pas skandinavus, o didžiausias – pietiečiams (Viduržemio jūros regiono gyventojams). Nepriklausomai nuo regiono, 677T alelio buvimas yra susijęs su homocisteino koncentracijos padidėjimu plazmoje; homozigotuose šis padidėjimas yra daug ryškesnis nei heterozigotų.
Didelis 677T alelio dažnis rodo, kad šios mutacijos nešėjai galėjo turėti tam tikrų pranašumų natūralioje atrankoje. Yra hipotezė, kad nevalgius sumažėjęs MTHFR aktyvumas sumažina homocisteino remetilinimą, taip išsaugant monokarboninius radikalus iš tetrahidrofolato metabolizmo gyvybiškai svarbiai DNR ir RNR sintezei. Remiantis kita hipoteze, mutantinio alelio nešiotojai rečiau suserga storosios žarnos vėžiu, dėl to mutacijos dažnis populiacijoje gali palaipsniui didėti.
677T mutacija ir vaisiaus nervinio vamzdelio defektai
677T mutacija skatina vidutinio sunkumo hiperhomocisteinemijos išsivystymą, ypač esant sumažėjusiam folio rūgšties kiekiui. Ši genetinio polinkio ir mitybos ypatybių sąveika padidina vaisiaus nervinio vamzdelio defektų atsiradimo riziką. Tyrimai parodė, kad 677T alelis dažniau aptinkamas tarp motinų, tėčių ir vaikų, kai vaisiui aptinkamas nervinio vamzdelio defektas. Nustatyta koreliacija tarp 677T alelio dažnio populiacijoje ir nervinio vamzdelio defektų dažnio.
Šiuo metu ryšys tarp vaisiaus nervinio vamzdelio defektų ir motinos homozigotiškumo 677T aleliui laikomas įrodytu. Tačiau nervinio vamzdelio defektų atsiradimas dėl žemos folatų būklės nėščioms moterims ne visada yra susijęs su 677T aleliu, o tai rodo tinkamo folio rūgšties vartojimo nėštumo metu svarbą. 677T alelio derinys su žema folio būkle kelia didesnę nervinio vamzdelio defektų atsiradimo riziką, nei esant vienam iš šių dviejų veiksnių.
677T mutacija ir kitos nėštumo komplikacijos
Moterys, turinčios 677TT genotipą, yra linkusios vystytis folio rūgšties vitamino trūkumui. Nėščioms moterims, kurioms šis alelis yra homozigotinis, folatų trūkumas gali būti nustatytas tik raudonuosiuose kraujo kūneliuose, o folatų koncentracija plazmoje gali būti neveikiama. Tačiau nėštumo metu homozigotinėms moterims folatų koncentracija sumažėja ne tik raudonųjų kraujo kūnelių viduje, bet ir kraujo plazmoje.
Tyrimai parodė, kad nėščioms moterims, sergančioms kraujagyslių ligomis, padidėja rizika susirgti nefropatija. Tai gerai sutampa su duomenimis apie didelės homocisteino koncentracijos kraujyje įtaką nėščių moterų nefropatijos išsivystymo rizikai. Be to, buvo įrodyta, kad homocisteino koncentracija kraujyje koreliuoja su fibronektino koncentracija ląstelėse, o tai rodo svarbų homocisteino vaidmenį endotelio disfunkcijos vystymuisi nėštumo metu. 677T alelio dažnio padidėjimas buvo pastebėtas ne tik vėlyvosios toksikozės (preeklampsijos), bet ir kitų nėštumo komplikacijų (placentos atsiskyrimo, vaisiaus augimo sulėtėjimo, priešgimdyminės vaisiaus mirties) atveju. 677T alelio derinys su kitais rizikos veiksniais padidina ankstyvo persileidimo riziką. Folio rūgšties įtraukimas į dietą žymiai sumažina nėštumo komplikacijų riziką. Profilaktinė folio rūgšties papildymo dieta reikšmė ypač ryški, kai yra hiperhomocisteinemija.
677T mutacija ir psichikos sutrikimai
Asmenys, kuriems yra sunkus MTHFR trūkumas, dažnai turi psichikos sutrikimų, kurie reaguoja į gydymą folio rūgštimi. Todėl yra hipotezė, kad 677T alelis yra susijęs su padidėjusia rizika susirgti šizofrenija, sunkiais depresiniais sutrikimais ir kitomis psichozėmis. Tačiau kol kas nėra gauta įtikinamų įrodymų, kad 677T alelis padidina psichikos ligų išsivystymo riziką. Tačiau negalima atmesti 677T alelio dalyvavimo psichikos sutrikimų vystyme kartu su kitais rizikos veiksniais.
Leideno mutacija
V krešėjimo faktoriaus geno Leideno mutacijai būdingas guanino nukleotido pakeitimas adenino nukleotidu 1691 padėtyje. Dėl to aminorūgštis argininas pakeičiama aminorūgštimi glutaminu 506 padėtyje baltymų grandinėje, kuri yra šio geno produktas. Prisiminkite, kad kiekvieną aminorūgštį koduoja trys DNR nukleotidai, vadinami kodonu. Todėl Leideno mutacija gali būti vadinama G1691A (guaninas į adeniną); Arg506Gln (argininas – glutaminas) arba R506Q (R yra vienos raidės arginino žymėjimas, Q – vienos raidės glutaminas). Visi trys pavadinimai yra tos pačios mutacijos sinonimai.
V krešėjimo faktoriaus genas yra 1 chromosomoje. Mutacija paveldima autosominiu dominuojančiu būdu. Tai reiškia, kad padidėjęs jautrumas trombozei, atsirandantis pakeitus R506Q, pasireiškia pakitusio geno buvimu tik vienoje pirmoje chromosomoje (kitoje pirmoje chromosomoje V faktoriaus genas nepakitęs). Ši būklė vadinama heterozigotiškumu. Leideno mutacija yra gana plačiai paplitusi populiacijoje. Vidutiniškai 4-6% Europos gyventojų yra heterozigotiniai nešiotojai. Leideno mutacijos (pakeisto geno abiejose pirmosiose chromosomose) homozigotinio nešiojimo atvejai populiacijoje yra labai reti.
Mutacija pavadinta Leidenu dėl to, kad Leideno trombofilijos tyrimų grupė pirmoji iššifravo genetinę kraujo krešėjimo sutrikimų, atsirandančių dėl šios mutacijos, prigimtį. Tai įvyko 1993 m.
V faktoriaus vaidmuo kraujo krešėjimo kaskadoje.
Kraujo krešėjimo faktorius V yra didelės molekulinės masės baltymas, kuris yra protrombinazės komplekso dalis. Protrombinazės kompleksas susidaro, kai kraujo krešėjimas suaktyvinamas išoriniu arba vidiniu keliu ir susideda iš aktyvuoto X faktoriaus (žymimo Xa), aktyvuoto faktoriaus V (žymimo Va) ir kalcio jonų, susijusių su fosfolipidų (PL) membranomis (dažniausiai trombocitų membranomis). Protrombinazės komplekso funkcija yra atskirti peptidų fragmentus iš protrombino molekulės, paverčiant protrombiną trombinu (fermentu, polimerizuojančiu fibriną iš fibrinogeno). Fibrinas yra galutinis kraujo krešėjimo produktas. Fermentas, skaidantis protrombiną protrombinazės komplekse, yra Xa faktorius, tačiau nedalyvaujant V faktoriui ši reakcija vyksta labai lėtai. Suaktyvintas V faktorius, fosfolipidų paviršiuje susijungęs su Xa, dešimtis tūkstančių kartų pagreitina trombino susidarymo reakciją. (žr. 3 pav.).
Kraujo krešėjimo ribojimas inaktyvuojant Va faktorių
Kraujo krešėjimo sistemos ypatybė yra daugybė teigiamų ir neigiamų grįžtamojo ryšio reakcijų. Darnus viso reakcijų komplekso derinys leidžia organizmui efektyviai susidoroti su kraujavimu ir užkirsti kelią kraujagyslių trombozei ten, kur nėra kraujavimo. Svarbi antikoaguliacijos kaskados dalis yra trombų susidarymo ribojimas dėl aktyvuoto baltymo C (lotyniška raidė C).
Pagrindinis krešėjimo fermentas, trombinas, yra vienas paslaptingiausių ir įdomiausių organizmo baltymų. Jis atlieka fermentinę funkciją, bet gali atlikti ir signalinės molekulės vaidmenį, dalyvaujant daugelyje kūno reakcijų, susijusių ne tik su trombų susidarymu. Trombinas, kaip fermentas, atlieka dvi tiesiogiai priešingas funkcijas: formuoja fibriną ir stabdo fibrino susidarymą. Trombinas įgyja savo antikoaguliacines savybes derinamas su trombomodulinu – endotelio (kraujagysles dengiančių ląstelių) membraniniu baltymu. Tuo pačiu metu trombino molekulė pakeičia savo konfigūraciją taip, kad nebegali dalyvauti krešėjimo reakcijoje, tačiau įgyja savybę skaidyti baltymą C – vieną iš nuo vitamino K priklausomų baltymų, sintetinamas kepenyse ir nuolat. esančios kraujyje. [1970-aisiais mokslininkai, tyrinėję nuo vitamino K priklausomus kepenų baltymus, juos pavadino lotyniškos abėcėlės raidėmis. Kitas nuo vitamino K priklausomas antikoaguliacijos kaskados baltymas yra aktyvuoto baltymo C baltymo S kofaktorius. Pastaruoju metu buvo paskelbta nedaug tyrimų su kitais šios serijos baltymais (baltymu Z ir baltymu M).]
Aktyvuotas baltymas C yra vienas pagrindinių fiziologinių antikoaguliantų, skaidančių aktyvuotus V ir VIII krešėjimo faktorius. Viena iš svarbių trombofilijos priežasčių yra šių veiksnių atsparumas žalingam APC poveikiui. Ši sąlyga vadinama APC atsparumu. Pagrindinė šio pasipriešinimo priežastis yra Leideno mutacija.
APC atsparumo priežastys Leideno mutacijoje
Normaliomis sąlygomis APC inaktyvuoja V faktorių, taip užkertant kelią jo įtraukimui į protrombinazės kompleksą. Norint inaktyvuoti Va faktorių aktyvuotu baltymu C, 506 padėtyje turi būti argininas. Arginino pakeitimas glutaminu lemia, kad faktorius V tampa atsparus APC skilimui. Be to, inaktyvuotas V faktorius yra būtinas, kad baltymo C/baltymo S kompleksas inaktyvuotų VIII krešėjimo faktorių. Todėl dėl nepakankamo inaktyvuoto faktoriaus V susidarymo susidaro aktyvuotas X faktorius, kuris yra protrombinazės dalis. komplekso, taip pat nustoja blokuoti aktyvuoto baltymo C. Taigi, organizme susidaro sąlygos, skatinančios protrombinazės komplekso hiperaktyvaciją, dėl kurios gali išsivystyti trombozė.
Normaliomis sąlygomis Leideno mutacijos nešiotojui trombozės gali nebūti. Trombozė išsivysto esant papildomiems rizikos veiksniams: nėštumas, hormoninių kontraceptikų vartojimas, padidėjęs homocisteino kiekis, MTHFR ir protrombino genų mutacijos, antifosfolipidiniai antikūnai. Svarbu pažymėti, kad pati homocisteinemija sukelia APC atsparumo vystymąsi, todėl šis derinys yra ypač pavojingas. Be to, Leideno mutacijos ir protrombino geno mutacijos G20210A derinys yra dažnesnis, nei būtų galima tikėtis iš atsitiktinio priskyrimo. Visa tai rodo pakankamai išsamaus paciento ištyrimo svarbą, jei įtariama trombofilinė būklė.
Leideno mutacija ir nėštumas
Leideno mutacijos buvimas padidina daugelio nėštumo komplikacijų atsiradimo tikimybę: ankstyvą nėštumo praradimą (rizika padidėja 3 kartus), vaisiaus vystymosi atsilikimą, vėlyvą toksikozę (preeklampsiją), vaisiaus placentos nepakankamumą. Dažniausiai Leideno mutaciją turinčioms moterims placentoje atsiranda trombozė, dėl kurios padidėja rizika susirgti visomis aukščiau išvardintomis komplikacijomis. Šių komplikacijų išsivystymo prevencija yra mažų aspirino dozių skyrimas, pradėtas dar prieš nėštumą, ir mažų dozių heparino preparatų (nefrakcionuoto heparino ir mažos molekulinės masės heparinų) injekcijos po oda. Šis gydymas yra saugus vaisiui ir gali žymiai sumažinti nepalankaus nėštumo baigties tikimybę.
Leideno mutacija ir hormoniniai kontraceptikai
Viena iš pavojingiausių hormoninių kontraceptikų komplikacijų yra trombozė ir tromboembolija. Paaiškėjo, kad daugelis moterų, turinčių tokias komplikacijas, yra heterozigotinės Leideno mutacijos nešiotojai. Vartojant hormoninius kontraceptikus, trombozės rizika padidėja 6-9 kartus. Jei pacientas turi Leideno mutaciją, trombozės atsiradimo rizika vartojant kontraceptikus padidėja 30-50 kartų. Todėl kai kurie autoriai siūlo, kad visos moterys, vartojančios hormoninius kontraceptikus arba planuojančios juos vartoti, turėtų būti patikrintos, ar nėra Leideno mutacijos.
Leideno mutacija ir chirurgija
Trombozė yra viena iš sunkiausių pooperacinio laikotarpio komplikacijų. Naujosios genetikos (genomikos) šalininkai siūlo ištirti, ar nėra Leideno mutacijos, visus pacientus, besiruošiančius didelėms operacijoms (gimdos mioma, cezario pjūvis, kiaušidžių cistos ir kt.).
Leideno mutacija ir vaisingumas
Neseniai atliktas tyrimas (Lancet, 2001 m. spalio 13 d.; 358 (9289): 1238-9) parodė, kad Leideno mutacijos nešiotojams embrionų pernešimas IVF yra maždaug 2 kartus didesnis nei pacientų, kurie šios mutacijos nenešioja. Šios įdomios išvados rodo, kad nepaisant padidėjusios komplikacijų tikimybės, pacientų, turinčių Leideno mutaciją, vaisingumo rodiklis gali būti didesnis (nėštumo tikimybė kiekviename cikle). Tai gali būti vienas iš paaiškinimų, kodėl ši mutacija taip plačiai paplito populiacijoje po jos atsiradimo maždaug prieš 20 tūkstančių metų. Veiksminga kraujagyslių trombozė implantacijos vietoje gali būti svarbi sėkmingų pirmųjų embriono ir gimdos gleivinės sąveikos etapų sąlyga. Beje, būtent todėl, gydant reprodukcinius sutrikimus, susijusius su trombofilija, embriono perkėlimo dienomis ir numatomomis implantacijos dienomis pernelyg didelė hipokoaguliacija nerekomenduojama.
Protrombino geno mutacija G20210A
Protrombino geno mutacijai G20210A būdingas guanino nukleotido pakeitimas adenino nukleotidu 20210 padėtyje. Mutaciją atrado Leideno trombofilijos tyrimų grupė 1996 m. Šios mutacijos ypatumas yra tas, kad nukleotidų pokytis yra 3 '-netransliuota sritis (sritis, esanti netransliuoto geno DNR sekos gale). Tai reiškia, kad pakeistos srities nukleotidų seka nedalyvauja koduojant protrombino geno aminorūgščių seką. Todėl esant šiai mutacijai, pačiame protrombine cheminių pokyčių nevyksta. Esant šiai mutacijai, aptinkamas padidėjęs chemiškai normalaus protrombino kiekis. Protrombino lygis gali būti pusantro ar du kartus didesnis nei normalus.
Protrombino genas yra vienuoliktoje chromosomoje. 2-3% Europos rasės atstovų yra heterozigotiniai geno nešiotojai. Homozigotinis mutacijos variantas yra labai retas radinys. Tarp afrikiečių ir mongoloidų rasės atstovų ši mutacija yra labai reta. Mutacija paveldima autosominiu dominuojančiu būdu. Tai reiškia, kad trombofilija pasireiškia net ir heterozigotiniam pakitusio geno nešiotojui.
Kai atsiranda trombozė, G20210A mutacija dažnai atsiranda kartu su Leideno mutacija. Ši mutacija yra visų su Leideno mutacija susijusių komplikacijų (persileidimas, vaisiaus ir placentos nepakankamumas, intrauterinė vaisiaus mirtis, preeklampsija, vaisiaus augimo sulėtėjimas, placentos atsiskyrimas) rizikos veiksnys.
Trombofilinės būklės (antifosfolipidinis sindromas, hiperhomocisteinemija, MTHFR, V faktoriaus ir protrombino genų mutacijos) yra viena iš svarbiausių persileidimo ir vaisiaus ir placentos nepakankamumo priežasčių. Ne nėštumo metu šios sąlygos gali sukelti trombozines hormoninių kontraceptikų ir chirurginių operacijų komplikacijas. Rekomenduojame atlikti molekulinį genetinį tyrimą šiais atvejais:
- jeigu praeityje ankstyvuoju nėštumo laikotarpiu buvo du ar daugiau vaisiaus augimo sustojimų;
- jeigu praeityje buvo sunkių nėštumo komplikacijų (sunkios vėlyvosios toksikozės formos, intrauterinė vaisiaus mirtis, vaisiaus augimo sulėtėjimas);
- jeigu giminaičiai turi trombozinių komplikacijų iki 50 metų (giliųjų venų trombozė, plaučių embolija, insultas, miokardo infarktas, staigi mirtis);
- su keliais nesėkmingais IVF bandymais;
- jei nustatomas padidėjęs antifosfolipidinių antikūnų kiekis ir (arba) padidėjęs homocisteino kiekis;
- planuojant ginekologines operacijas;
- planuojant hormoninę kontracepciją.
Beje, įvairiose vietose gali būti mutacijų.
Kai mutantinis MTHFR genas aptinkamas heterozigotinėje būsenoje*, nėra jokios įtikinamos priežasties baimintis. Norint išvengti hiperkoaguliacijos būklių, nėštumo metu rekomenduojama kasdien gerti po 0,4 mg folio rūgšties per dvi dozes, gerai maitintis ir kartą per tris mėnesius (arba pagal indikacijas) tirti hemostazogramą.
Dažniausias fermento defektas, susijęs su vidutiniu HC (homocisteino) koncentracijos padidėjimu, yra geno, koduojančio MTHFR, mutacija. MTHFR katalizuoja folio rūgšties pavertimą aktyvia forma. Iki šiol buvo aprašytos 9 MTHFR geno, esančio 1p36.3 lokuse, mutacijos. Dažniausias iš jų yra C677T pakeitimas (MTHFR baltyme – valino pakeitimas alaninu), pasireiškiantis termolabilumu ir MTHFR fermento aktyvumo sumažėjimu. Pastebėta, kad padidinus folatų kiekį maiste, galima išvengti GC koncentracijos padidėjimo plazmoje.
Homocisteino kiekio padidėjimas kraujo plazmoje tiesiogiai koreliuoja su trombomodulino sintezės slopinimu, AT-III ir endogeninio heparino aktyvumo sumažėjimu, taip pat su tromboksano A2 gamybos aktyvavimu. Ateityje tokie pokyčiai sukelia mikrotrombozę ir mikrocirkuliacijos sutrikimus, o tai savo ruožtu vaidina svarbų vaidmenį spiralinių arterijų patologijoje ir akušerinių komplikacijų, susijusių su gimdos placentos kraujotakos pokyčiais, vystymuisi. nuoroda
Padidėjusio homocisteino kiekio kraujyje priežastis: MTHFR geno C677T variantas - fermento metilentetrahidrofolato reduktazės geno mutacija.
Citoziną pakeitus timinu 677 padėtyje, fermento funkcinis aktyvumas sumažėja iki 35% vidutinės vertės.
Polimorfizmo duomenys:
*homozigotų atsiradimo dažnis populiacijoje yra 10-12 proc.
*heterozigotų atsiradimo populiacijoje dažnis – 40 proc.
T varianto nešiotojai nėštumo metu patiria folio rūgšties trūkumą, o tai lemia vaisiaus nervinio vamzdelio vystymosi defektus.
Rūkymas padidina 677T varianto poveikį.
Folio rūgšties vartojimas gali žymiai sumažinti šio polimorfizmo pasekmių riziką.
Apskritai, kas kur bus nuvežtas... Tiksliai pasakyti neįmanoma. Nuo tėvo taip pat priklauso, kas yra jo genome.
Pabandykite užduoti savo klausimą išsamiau čia - nuoroda
Viskas yra Dievo galioje. Čia statistika bejėgė.
Heterozigotinės mutacijos būsena
Padėk man, prašau.
Tiesioginis automatinis sekos nustatymas buvo naudojamas Notch 3 geno (Cadasil sindromo) mutacijų analizei.
Aptikta mutacija c.268C T, Arg90Cys heterozigotinėje būsenoje, aprašyta HGMD mutacijų duomenų bazėje.
Iš anksto dėkoju!
Taip pat nepamirškite padėkoti savo gydytojams.
genetikas7 22:07
reikia žinoti dėl ko buvo atlikta ekspertizė, kas jam vadovavo ir pamatyti išvadą.
Apžiūros priežastis buvo mano būklė, kai atvykau į kliniką. Man staiga atsirado silpnumas ir praradau kalbą. Kazanėje praėjau visus įmanomus testus ir egzaminus. Nustatyta: progresuojanti leukoencefalopatija, tikriausiai sukelta izoliuoto smegenų vaskulito, pasireiškianti vidutinio sunkumo pažinimo sutrikimais, bulbariniu sindromu, piramidės nepakankamumu. Hiperhomocisteinemija. Hipercholesterolemija. Profesorius rekomendavo atlikti molekulinę genetinę Notch-3 geno mutacijos diagnozę.
Molekulinės genetinės laboratorijos išvadą jau išsiunčiau ankstesniame laiške.
Daktare, padėk man, prašau! Iššifruokite šią išvadą.
Analizė patvirtino sindromą, kurį įtarė gydytojas.
Labai ačiū už atsakymą. Dabar aš žinau, kad sergu. Kol liga mane visiškai neužvaldė. Matyt, tai ateis vėliau. Na, toks mano likimas.
Vis tiek norėčiau sužinoti, kas yra heterozigotinė mutacija. Akivaizdu, kad tai kažkaip atsispindi ligos paveldėjimo principe. Turiu du vaikus, berniukus. Mano sesuo turi dvi mergaites. Ji už mane jaunesnė, jai 38 metai. Man 44 metai. Liga paveldėjau iš savo tėvo. Jis mirė sulaukęs 61 metų. Mirties priežastis buvo insultas. Jo jaunesnis brolis ir vyresnioji sesuo gyvi ir palyginti sveiki. Jų vaikai taip pat sveiki. Ar tikrai aš vienintelis turiu mutaciją?
Jei atsakysite bent į kelis iš šių klausimų, būsiu jums labai dėkingas.
Viskas kas geriausia.
genetikas3 10:35
Ta pati galimybė egzistavo ir tau, ir tavo seseriai. Kadangi ji yra jaunesnė už jus, kol kas nežinoma, ar ji tai paveldėjo.
Jūsų sesuo ir jūsų vaikai gali atlikti tą patį genetinį tyrimą, kuris buvo atliktas jums. Jei jie nori dabar išsiaiškinti, ar jie paveldėjo mutaciją, ar ne.
Heterozigotinė mutacija, ką tai reiškia?
Homozigotiškumas ir heterozigotiškumas, dominavimas ir recesyvumas.
Homozigotiškumas (iš graikų kalbos „homo“ lygus, „zigota“ apvaisintas kiaušinis) yra diploidinis organizmas (arba ląstelė), turintis identiškus alelius homologinėse chromosomose.
Gregoras Mendelis pirmasis nustatė faktą, rodantį, kad išvaizdos panašūs augalai gali smarkiai skirtis paveldimomis savybėmis. Asmenys, kurie neskyla kitoje kartoje, vadinami homozigotais. Asmenys, kurių palikuonys pasižymi charakterių skilimu, vadinami heterozigotiniais.
Homozigotiškumas – tai paveldimo organizmo aparato būsena, kurioje homologinės chromosomos turi tą pačią tam tikro geno formą. Geno perėjimas į homozigotinę būseną lemia recesyvinių alelių pasireiškimą organizmo struktūroje ir funkcijose (fenotipas), kurių poveikį, esant heterozigotiškumui, slopina dominuojantys aleliai. Homozigotiškumo testas yra segregacijos nebuvimas tam tikrų rūšių kryžminimo metu. Homozigotinis organizmas tam tikram genui gamina tik vieno tipo gametas.
Heterozigotiškumas yra bet kuriam hibridiniam organizmui būdinga būklė, kai jo homologinės chromosomos turi skirtingas konkretaus geno formas (alelius) arba skiriasi santykine genų padėtimi. Terminą „heterozigotiškumas“ pirmą kartą įvedė anglų genetikas W. Batesonas 1902 m. Heterozigotiškumas atsiranda, kai skirtingos genetinės ar struktūrinės sudėties gametos susilieja į heterozigotą. Struktūrinis heterozigotiškumas atsiranda, kai įvyksta vienos iš homologinių chromosomų chromosomų persitvarkymas; jį galima rasti mejozės arba mitozės metu. Heterozigotiškumas atskleidžiamas naudojant bandomąjį kryžminimą. Heterozigotiškumas, kaip taisyklė, yra seksualinio proceso pasekmė, tačiau gali atsirasti dėl mutacijos. Esant heterozigotiškumui, kenksmingų ir mirtinų recesyvinių alelių poveikį slopina atitinkamas dominuojantis alelis ir atsiranda tik tada, kai šis genas pereina į homozigotinę būseną. Todėl heterozigotiškumas yra plačiai paplitęs natūraliose populiacijose ir, matyt, yra viena iš heterozės priežasčių. Heterozigotiškumo dominuojančių alelių maskavimo efektas yra žalingų recesyvinių alelių išlikimo ir plitimo populiacijoje priežastis (vadinamasis heterozigotinis vežimas). Jų identifikavimas (pavyzdžiui, tiriant tėvus pagal palikuonis) atliekamas atliekant bet kokius veisimo ir selekcijos darbus, taip pat atliekant medicinines ir genetines prognozes.
Savo žodžiais galime teigti, kad veisimo praktikoje homozigotinė genų būsena vadinama „teisinga“. Jei abu požymį kontroliuojantys aleliai yra vienodi, tada gyvūnas vadinamas homozigotu ir veisdamas jis paveldės šią konkrečią savybę. Jei vienas alelis yra dominuojantis, o kitas – recesyvinis, gyvūnas vadinamas heterozigotiniu ir išoriškai parodys dominuojančią savybę, tačiau paveldės arba dominuojančią, arba recesyvinę.
Bet kuris gyvas organizmas turi DNR (dezoksiribonukleino rūgšties) molekulių skyrių, vadinamą chromosomomis. Dauginimosi metu lytinės ląstelės kopijuoja paveldimą informaciją savo nešikliais (genais), kurie sudaro spiralės formos chromosomų sekciją, esančią ląstelių viduje. Genai, esantys tuose pačiuose homologinių chromosomų lokusuose (griežtai apibrėžtose padėtyse chromosomoje) ir lemiantys bet kurio požymio išsivystymą, vadinami aleliniais. Diploidiniame (dvigubo, somatinio) rinkinyje dvi homologinės (identiškos) chromosomos ir atitinkamai du genai turi šių skirtingų savybių vystymąsi. Vienų bruožų vyravimas kito atžvilgiu vadinamas dominavimu, o genai – dominuojantys. Požymis, kurio pasireiškimas yra slopinamas, vadinamas recesyviniu. Alelio homozigotiškumas yra dviejų identiškų genų (paveldimos informacijos nešėjų) buvimas jame: arba du dominuojantys, arba du recesyviniai. Alelio heterozigotiškumas – tai dviejų skirtingų genų buvimas jame, t.y. vienas iš jų yra dominuojantis, o kitas – recesyvinis. Aleliai, kurie heterozigote suteikia tokį patį bet kurio paveldimo požymio pasireiškimą kaip ir homozigote, vadinami dominuojančiais. Aleliai, kurie pasireiškia tik homozigote, bet yra nematomi heterozigote arba yra slopinami veikiant kitam dominuojančiam aleliui, vadinami recesyviniais.
Homozigotiškumo, heterozigotiškumo ir kitų genetikos pagrindų principus pirmasis suformulavo genetikos įkūrėjas abatas Gregoras Mendelis savo trijų paveldėjimo dėsnių pavidalu.
Pirmasis Mendelio dėsnis: „Palikuonys, sukryžminus individus, homozigotinius skirtingoms to paties geno alėjoms, yra vienodo fenotipo ir heterozigotinio genotipo“.
Antrasis Mendelio dėsnis: „Kryžminus heterozigotines formas, stebimas natūralus palikuonių skilimas santykiu 3:1 fenotipo ir 1:2:1 genotipo atžvilgiu.
Trečiasis Mendelio dėsnis: „Kiekvieno geno aleliai yra paveldimi nepriklausomai nuo gyvūno kūno sudėties.
Šiuolaikinės genetikos požiūriu jo hipotezės atrodo taip:
1. Kiekvieną tam tikro organizmo požymį valdo alelių pora. Individas, gavęs vienodus alelius iš abiejų tėvų, vadinamas homozigotiniu ir žymimas dviem identiškomis raidėmis (pavyzdžiui, AA arba aa), o jei gauna skirtingas – heterozigotiniu (Aa).
2. Jeigu organizme yra du skirtingi tam tikro požymio aleliai, tai vienas iš jų (dominuojantis) gali pasireikšti, visiškai nuslopindamas kito pasireiškimą (recesyvinis). (Pirmosios kartos palikuonių dominavimo arba vienodumo principas). Kaip pavyzdį paimkime monohibridinį (tik pagal spalvą) kryžminimą tarp kokerių. Tarkime, kad abu tėvai yra homozigotiniai pagal spalvą, todėl juodas šuo turės genotipą, kurį žymėsime, pavyzdžiui, AA, o gelsvos spalvos šuo turės aa. Abu individai gamins tik vieno tipo lytines ląsteles: juodoji tik A, o gelsvai gelsvai tik a. Kad ir kiek šuniukų gimtų tokioje vadoje, jie visi bus juodi, nes juoda spalva dominuoja. Kita vertus, jie visi bus gelsvos spalvos geno nešiotojai, nes jų genotipas yra Aa. Tiems, kurie nėra pernelyg aiškūs, atkreipkite dėmesį, kad recesyvinis bruožas (šiuo atveju gelsva spalva) pasireiškia tik homozigotinėje būsenoje!
3. Kiekviena lytinė ląstelė (gameta) gauna po vieną iš kiekvienos alelių poros. (Skaldymo principas). Jei sukryžminsime pirmosios kartos palikuonis arba bet kuriuos du kokerius, turinčius Aa genotipą, antros kartos palikuoniuose bus stebimas skilimas: Aa + aa = AA, 2Aa, aa. Taigi, fenotipinis padalijimas atrodys kaip 3:1, o genotipinis padalijimas atrodys kaip 1:2:1. Tai reiškia, kad poruodami du juodus heterozigotinius kokerius galime turėti 1/4 tikimybę turėti juodus homozigotinius šunis (AA), 2/4 galimybę turėti juodus heterozigotus (Aa) ir 1/4 galimybę turėti gelsvos spalvos šunis (aa). . Gyvenimas nėra toks paprastas. Kartais du juodi heterozigotiniai kokeriai gali pagimdyti gelsvos spalvos šuniukus arba gali būti visi juodi. Mes tiesiog apskaičiuojame tikimybę, kad tam tikras požymis atsiras šuniukams, o ar jis pasireikš, priklauso nuo to, kurie aleliai atsidūrė apvaisintuose kiaušinėliuose.
4. Formuojantis gametoms, bet kuris alelis iš vienos poros gali patekti į kiekvieną iš jų kartu su bet kuriuo kitu iš kitos poros. (Nepriklausomo paskirstymo principas). Daugelis bruožų yra paveldimi nepriklausomai, pavyzdžiui, akių spalva gali priklausyti nuo bendros šuns spalvos, tačiau tai praktiškai neturi nieko bendra su ausų ilgiu. Jei imsime dihibridinį kryžių (dviem skirtingiems požymiams), pamatysime tokį santykį: 9: 3: 3: 1
5. Kiekvienas alelis perduodamas iš kartos į kartą kaip atskiras, nekintantis vienetas.
b. Kiekvienas organizmas paveldi po vieną alelį (kiekvienam požymiui) iš kiekvieno iš tėvų.
Konkrečiam genui, jei du individo nešiojami aleliai yra vienodi, kuris iš jų vyraus? Kadangi dėl alelių mutacijos dažnai prarandama funkcija (tušti aleliai), individas, turintis tik vieną tokį alelį, turės ir „normalų“ (laukinio tipo) alelį tam pačiam genui; normaliai funkcijai palaikyti dažnai pakaks vienos įprastos kopijos. Kaip analogiją įsivaizduokime, kad statome mūrinę sieną, bet vienas iš dviejų nuolatinių rangovų streikuoja. Kol likęs tiekėjas gali tiekti mums pakankamai plytų, mes galime toliau statyti savo sieną. Genetikai šį reiškinį, kai vienas iš dviejų genų dar gali užtikrinti normalią funkciją, vadina dominavimu. Nustatyta, kad normalus alelis dominuoja nenormalaus alelio atžvilgiu. (Kitaip tariant, galime sakyti, kad neteisingas alelis yra recesyvinis normaliam aleliui.)
Kai kalbama apie genetinę anomaliją, kurią „neša“ individas ar giminė, tai reiškia, kad yra mutavęs genas, kuris yra recesyvus. Nebent atliksime sudėtingų testų, skirtų tiesiogiai aptikti šį geną, negalėsime vizualiai identifikuoti nešiotojo iš asmens, turinčio dvi normalias geno kopijas (alelius). Deja, nesant tokio tyrimo, kurjeris nebus laiku aptiktas ir neišvengiamai perduos mutacijos alelį kai kuriems savo palikuonims. Kiekvienas asmuo gali būti panašiai „užbaigtas“ ir turėti keletą šių tamsių paslapčių savo genetiniame bagaže (genotipas). Tačiau mes visi turime tūkstančius skirtingų genų, skirtų daugeliui skirtingų funkcijų, ir nors šie anomalijos yra reti, tikimybė, kad du nesusiję individai, turintys tą patį „nenormalumą“, susitiks, kad galėtų daugintis, yra labai maža.
Kartais asmenys, turintys vieną normalų alelį, gali turėti „tarpinį“ fenotipą. Pavyzdžiui, Basenji, turintis vieną alelį dėl piruvatkinazės trūkumo (fermento trūkumas, sukeliantis lengvą anemiją), vidutinė raudonųjų kraujo kūnelių gyvenimo trukmė yra 12 dienų. Tai yra tarpinis tipas tarp įprasto 16 dienų ciklo ir 6,5 dienų ciklo šuniui su dviem neteisingais aleliais. Nors tai dažnai vadinama nepilnu dominavimu, šiuo atveju būtų geriau sakyti, kad dominavimo iš viso nėra.
Paimkime mūsų plytų sienos analogiją šiek tiek toliau. Ką daryti, jei vieno plytų tiekimo neužtenka? Mums liks žemesnė (arba trumpesnė) siena, nei tikėtasi. Ar tai bus svarbu? Tai priklauso nuo to, ką norime daryti su „siena“ ir galbūt nuo genetinių veiksnių. Rezultatas gali būti ne toks pat dviem sieną pastačiusiems žmonėms. (Žema siena gali užkirsti kelią potvyniui, bet ne potvyniui!) Jei įmanoma, kad asmuo, turintis tik vieną neteisingo alelio kopiją, išreikš jį netinkamu fenotipu, tada tas alelis turėtų būti laikomas dominuojančiu. Jos atsisakymas visada tai daryti apibrėžiamas terminu „penetrance“.
Trečia galimybė – vienas iš rangovų mums tiekia plytas pagal užsakymą. To nesuprasdami dirbame toliau – galiausiai siena griūva. Galima sakyti, kad vyrauja brokuotos plytos. Pažanga suprantant keletą dominuojančių žmonių genetinių ligų rodo, kad tai yra pagrįsta analogija. Dauguma dominuojančių mutacijų paveikia baltymus, kurie yra didelių makromolekulinių kompleksų komponentai. Dėl šių mutacijų pasikeičia baltymai, kurie negali tinkamai sąveikauti su kitais komponentais, todėl sugenda visas kompleksas (sugedusios plytos – nukritusi siena). Kiti yra reguliavimo sekose, esančiose greta genų, todėl genas transkribuojamas netinkamu laiku ir vietoje.
Dominuojančios mutacijos gali išlikti populiacijose, jei jų sukeliamos problemos yra subtilios ir ne visada ryškios arba atsiranda vėlyvoje gyvenimo stadijoje, kai paveiktas individas dalyvauja dauginimosi procese.
Recesyvinis genas (t. y. jo nustatytas bruožas) gali nepasireikšti per vieną ar kelias kartas, kol nesusidursite su dviem vienodais recesyviniais genais iš kiekvieno iš tėvų (staigus tokio požymio pasireiškimas palikuonims neturėtų būti painiojamas su mutacija).
Šunys, turintys tik vieną recesyvinį geną – bet kurio požymio determinantą – nerodys šio bruožo, nes recesyvinio geno poveikis bus užmaskuotas jo suporuoto dominuojančio geno įtakos pasireiškimu. Tokie šunys (recesyvinio geno nešiotojai) gali būti pavojingi veislei, jei šis genas nulems nepageidaujamo požymio atsiradimą, nes jį perduos savo palikuonims, o jie vėliau išsaugos veislei. Jei netyčia ar neapgalvotai suporuosite du tokio geno nešiotojus, jie susilauks nepageidautinų savybių turinčių palikuonių.
Dominuojančio geno buvimas visada aiškiai ir išoriškai pasireiškia atitinkamu ženklu. Todėl dominuojantys genai, turintys nepageidaujamą požymį, veisėjui kelia daug mažesnį pavojų nei recesyviniai, nes jų buvimas visada pasireiškia, net jei dominuojantis genas „dirba“ be partnerio (Aa).
Tačiau, matyt, viską apsunkina tai, kad ne visi genai yra visiškai dominuojantys ar recesyviniai. Kitaip tariant, vieni dominuoja labiau nei kiti ir atvirkščiai. Pavyzdžiui, kai kurie veiksniai, lemiantys kailio spalvą, gali būti dominuojantys, bet vis tiek nepasirodo išoriškai, nebent juos palaiko kiti genai, kartais net recesyviniai.
Poravimosi santykiai ne visada tiksliai atitinka tikėtinus vidutinius rezultatus ir norint gauti patikimą rezultatą iš tam tikro kergimo, reikia išvesti didelę vada arba daug palikuonių keliose vadose.
Kai kurios išorinės savybės gali būti „dominuojančios“ kai kuriose veislėse, o „recesyvinės“ kitose. Kiti bruožai gali atsirasti dėl kelių genų arba pusgenų, kurie nėra paprasti Mendelio dominuojantys ar recesyviniai.
Genetinių sutrikimų diagnostika
Genetinių sutrikimų diagnozė kaip genetinių ligų atpažinimo ir įvardijimo doktrina daugiausia susideda iš dviejų dalių
patologinių požymių, tai yra atskirų asmenų fenotipinių nukrypimų, nustatymas; aptiktų nukrypimų paveldimumo įrodymas. Terminas „genetinis sveikatos įvertinimas“ reiškia fenotipiškai normalaus individo tyrimą, siekiant nustatyti nepalankius recesyvinius alelius (heterozigotiškumo testas). Kartu su genetiniais metodais naudojami ir metodai, kurie pašalina aplinkos poveikį. Įprasti tyrimo metodai: vertinimas, laboratorinė diagnostika, patologinės anatomijos, histologijos ir patofiziologijos metodai. Ypatingi metodai labai svarbūs yra citogenetiniai ir imunogenetiniai metodai. Ląstelių kultūros metodas prisidėjo prie didelės pažangos diagnozuojant ir atliekant genetinę paveldimų ligų analizę. Per trumpą laiką šis metodas leido jo pagalba ištirti apie 20 genetinių defektų, rastų žmonėms (Rerabek ir Rerabek, 1960; New, 1956; Rapoport, 1969), daugeliu atvejų galima atskirti homozigotus nuo heterozigotai, turintys recesyvinį paveldėjimo tipą
Imunogenetiniai metodai naudojami tiriant kraujo grupes, serumo ir pieno baltymus, sėklinių skysčių baltymus, hemoglobino tipus ir kt. Daugelio baltymų lokusų su daugybe alelių atradimas atvedė prie Mendelio genetikos „Renesanso eros“. Baltymų lokusai naudojami:
nustatyti atskirų gyvūnų genotipą
tiriant tam tikrus specifinius defektus (imunoparezę)
ryšio tyrimams (žymenų genai)
genų nesuderinamumo analizei
aptikti mozaikiškumą ir chimerizmą
Defekto buvimas nuo gimimo momento, defektai, atsirandantys tam tikrose linijose ir darželiuose, bendro protėvio buvimas kiekvienu anomaliniu atveju nereiškia tam tikros būklės ir genetinės prigimties paveldimumo. Nustačius patologiją, būtina gauti jos genetinės priežasties įrodymus ir nustatyti paveldėjimo tipą. Taip pat būtinas statistinis medžiagos apdorojimas. Genetinė ir statistinė analizė atliekama dviem duomenų grupėms:
Populiacijos duomenys – įgimtų anomalijų dažnis visoje populiacijoje, įgimtų anomalijų dažnis subpopuliacijoje
Šeimos duomenys – genetinio determinacijos ir paveldėjimo tipo, giminystės koeficientų ir protėvių koncentracijos laipsnio nustatymo įrodymai.
Tiriant genetinį sąlygotumą ir paveldėjimo tipą, to paties (teoriškai) genotipo tėvų grupės palikuonių normalių ir defektinių fenotipų skaitiniai santykiai lyginami su segregacijos koeficientais, apskaičiuotais pagal dvinario tikimybę pagal Mendelio metodą. įstatymai. Norint gauti statistinę medžiagą, reikia apskaičiuoti paveiktų ir sveikų asmenų dažnį tarp probando kraujo giminaičių per kelias kartas, nustatyti skaitinį santykį sujungus atskirus duomenis ir apjungti duomenis apie mažas šeimas su atitinkamai identiškais tėvų genotipais. Taip pat svarbi informacija apie vados dydį ir šuniukų lytį (siekiant įvertinti susieto ar lytimi ribojamo paveldimumo galimybę).
Tokiu atveju būtina rinkti atrankos duomenis:
Kompleksinė atranka – atsitiktinė tėvų atranka (naudojama tikrinant dominuojančią savybę)
Tikslinga atranka – visi šunys, turintys „blogų“ bruožų populiacijoje, nuodugniai ištyrus
Individuali atranka – anomalijos atsiradimo tikimybė tokia maža, kad ji įvyksta vienam šuniukui iš vados
Daugkartinė atranka yra tarpinė tarp tikslinio ir individualaus, kai vadoje yra daugiau nei vienas paveiktas šuniukas, tačiau ne visi jie yra zonduoti.
Visi metodai, išskyrus pirmąjį, neleidžia kergti Nn genotipo šunų, kurie nesukelia anomalijų vadose. Yra įvairių būdų taisyti duomenis: N.T.J. Bailey (79), L. L. Kawaii-Sforza ir W. F. Bodme ir K. Stehr.
Genetinis populiacijos apibūdinimas prasideda nuo tiriamos ligos ar bruožo paplitimo įvertinimo. Remiantis šiais duomenimis, nustatomi genų ir atitinkamų genotipų dažniai populiacijoje. Populiacijos metodas leidžia ištirti atskirų genų ar chromosomų anomalijų pasiskirstymą populiacijose. Norint išanalizuoti populiacijos genetinę struktūrą, būtina ištirti didelę individų grupę, kuri turi būti reprezentatyvi, leidžianti spręsti apie populiaciją kaip visumą. Šis metodas yra informatyvus tiriant įvairias paveldimos patologijos formas. Pagrindinis paveldimų anomalijų tipo nustatymo metodas yra giminingų asmenų grupių kilmės dokumentų, kuriuose tiriamos ligos atvejai buvo užfiksuoti pagal šį algoritmą, analizė:
Anomalių gyvūnų kilmės nustatymas naudojant veisimo korteles;
Anomalių individų kilmės dokumentų sudarymas, siekiant ieškoti bendrų protėvių;
Anomalijos paveldėjimo tipo analizė;
Atlikti genetinius ir statistinius anomalijos atsiradimo atsitiktinumo laipsnio ir pasireiškimo populiacijoje dažnio skaičiavimus.
Genealoginis kilmės dokumentų analizės metodas užima pirmaujančią vietą lėtai besidauginančių gyvūnų ir žmonių genetiniuose tyrimuose. Ištyrus kelių kartų giminaičių fenotipus, galima nustatyti bruožo paveldėjimo pobūdį ir atskirų šeimos narių genotipus, nustatyti pasireiškimo tikimybę ir palikuonių rizikos laipsnį susirgti konkrečia liga.
Nustatant paveldimą ligą, atkreipiamas dėmesys į tipinius genetinio polinkio požymius. Patologija dažniau pasireiškia giminingų gyvūnų grupėje nei visoje populiacijoje. Tai padeda atskirti įgimtą ligą nuo veislės polinkio. Tačiau kilmės dokumentų analizė rodo, kad yra šeimyninių ligos atvejų, o tai rodo, kad yra tam tikras genas ar genų grupė, atsakinga už tai. Antra, paveldimas defektas dažnai paveikia tą pačią anatominę sritį giminingų gyvūnų grupėje. Trečia, su giminingumu, yra daugiau ligos atvejų. Ketvirta, paveldimos ligos dažnai pasireiškia anksti ir dažnai būna pastovaus amžiaus.
Genetinės ligos dažniausiai paveikia kelis vados gyvūnus, priešingai nei intoksikacijos ir infekcinės ligos, kurios paveikia visą vados. Įgimtos ligos labai įvairios – nuo santykinai gerybinių iki mirtinų. Jų diagnozė dažniausiai grindžiama anamneze, klinikiniais požymiais, giminingų gyvūnų ligos istorija, bandymų kryžminimo rezultatais ir tam tikrais diagnostiniais tyrimais.
Nemaža dalis monogeninių ligų yra paveldimos recesyviniu būdu. Tai reiškia, kad esant autosominei atitinkamo geno lokalizacijai, paveikiami tik homozigotinių mutacijų nešėjai. Mutacijos dažniausiai būna recesyvinės ir pasireiškia tik homozigotinėje būsenoje. Heterozigotai yra kliniškai sveiki, tačiau jie taip pat gali perduoti savo vaikams mutantinį arba normalų geno variantą. Taigi per ilgą laiką latentinė mutacija gali būti perduodama iš kartos į kartą. Esant autosominiam recesyviniam paveldėjimo tipui sunkiai sergančių pacientų, kurie neišgyvena iki reprodukcinio amžiaus arba kurių dauginimosi potencialas yra smarkiai sumažėjęs, kilmės dokumentuose, retai įmanoma nustatyti sergančius giminaičius, ypač kylančioje linijoje. Išimtis yra šeimos, kuriose yra aukštas giminystės lygis.
Šunys, turintys tik vieną recesyvinį geną – bet kurio požymio determinantą – nerodys šio bruožo, nes recesyvinio geno poveikis bus užmaskuotas jo suporuoto dominuojančio geno įtakos pasireiškimu. Tokie šunys (recesyvinio geno nešiotojai) gali būti pavojingi veislei, jei šis genas nulems nepageidaujamo požymio atsiradimą, nes jį perduos savo palikuonims. Jei du tokio geno nešiotojai atsitiktinai ar sąmoningai suporuojami, jie susilauks palikuonių su nepageidaujamomis savybėmis.
Numatomas palikuonių pasiskirstymo pagal vieną ar kitą požymį santykis maždaug pateisinamas, kai vada yra ne mažesnė kaip 16 šuniukų. Įprasto dydžio šuniukų vadai galime kalbėti tik apie didesnę ar mažesnę recesyvinio geno nulemto požymio pasireiškimo tikimybę tam tikros žinomo genotipo patelių poros palikuonims.
Atranka dėl recesyvinių anomalijų gali būti atliekama dviem būdais. Pirmasis iš jų yra pašalinti iš veisimo šunis, turinčius anomalijų, t. y. homozigotų. Anomalijos atsiradimas su tokia atranka pirmosiose kartose smarkiai mažėja, o vėliau lėčiau, išlikdamas santykinai žemame lygyje. Priežastis, dėl kurios kai kurios anomalijos net ir ilgos ir nuolatinės atrankos metu visiškai pašalinamos, yra, pirma, daug lėtesnis recesyvinių genų nešiotojų skaičiaus sumažėjimas nei homozigotų. Antra, kai mutacijos šiek tiek nukrypsta nuo normos, veisėjai ne visada naikina nenormalius šunis ir nešiotojus.
Su autosominiu recesyviniu paveldėjimo tipu:
Požymis gali būti perduodamas iš kartos į kartą net ir turint pakankamai palikuonių
Šis simptomas gali pasireikšti vaikams, jei jo (akivaizdžiai) nėra tėvams. Tada jis nustatomas 25% atvejų vaikams
Požymį paveldi visi vaikai, jei serga abu tėvai
Šis simptomas pasireiškia 50% vaikų, jei vienas iš tėvų serga
Vyriški ir moteriški palikuonys šią savybę paveldi vienodai
Taigi, absoliučiai visiškai pašalinti anomaliją iš esmės įmanoma, jei bus nustatyti visi nešiotojai. Tokio aptikimo schema: recesyvinių mutacijų heterozigotai kai kuriais atvejais gali būti aptikti laboratoriniais tyrimo metodais. Tačiau norint genetiškai identifikuoti heterozigotinius nešiotojus, būtina atlikti analitinį kryžminimą – įtariamo šuns nešiotojo poravimą su homozigotiniu nenormaliu (jei anomalija nežymiai paveikia organizmą) arba su anksčiau nustatytu nešiotoju. Jei dėl tokių kryžminimo, be kita ko, gimsta nenormalūs šuniukai, tirtas patelis aiškiai identifikuojamas kaip nešiotojas. Tačiau jei tokie šuniukai nenustatyti, tai iš gautos ribotos šuniukų imties negalima daryti vienareikšmiškos išvados. Tikimybė, kad toks patelis yra nešiotojas, mažėja plečiant imtį – daugėja normalių šuniukų, gimusių po poravimosi su juo.
Sankt Peterburgo Veterinarijos akademijos katedroje atlikta šunų genetinės apkrovos struktūros analizė ir nustatyta, kad didžiausia dalis – 46,7 proc. – yra monogeninio autosominio recesyvinio tipo paveldimos anomalijos; anomalijos su visišku dominavimu siekė 14,5 %; 2,7 % anomalijų pasirodė kaip nepilni dominuojantys bruožai; 6,5% anomalijų yra paveldimos su lytimi, 11,3% paveldimų požymių turi poligeninį paveldėjimo tipą ir 18% 3% viso paveldimų anomalijų spektro, paveldėjimo tipas nenustatytas. Bendras šunų anomalijų ir ligų, turinčių paveldimą pagrindą, skaičius buvo 186 vienetai.
Kartu su tradiciniais atrankos ir genetinės prevencijos metodais aktualus yra ir fenotipinių mutacijų žymenų naudojimas.
Genetinių ligų stebėjimas yra tiesioginis nesergančių tėvų palikuonių paveldimų ligų vertinimo metodas. „Sargybos“ fenotipai gali būti: gomurio plyšimas, lūpos plyšimas, kirkšnies ir bambos išvaržos, naujagimių hidrocelė, naujagimių šuniukų traukuliai. Sergant monogeninėmis fiksuotomis ligomis, galima nustatyti tikrąjį nešiotojas per su juo susijusį žymeklio geną.
Esama šunų veislinė įvairovė suteikia unikalią galimybę ištirti daugelio morfologinių požymių genetinę kontrolę, kurių įvairios kombinacijos lemia veislės standartus. Tokią situaciją galima iliustruoti dviem iš šiuo metu egzistuojančių naminių šunų veislių, kurios viena nuo kitos skiriasi bent jau tokiomis morfologinėmis savybėmis kaip ūgis ir svoris. Viena vertus, tai yra anglų mastifų veislė, kurios atstovai pasiekia 80 cm ūgį ties ketera ir sveria daugiau nei 100 kg, o Chi Hua Hua veislė - 30 cm ir 2,5 kg.
Prijaukinimo procesas apima gyvūnų atranką pagal jų išskirtines savybes, žmogaus požiūriu. Laikui bėgant, kai šuo buvo pradėtas laikyti kaip kompanionas ir dėl jo estetinės išvaizdos, selekcijos kryptis pasikeitė – buvo auginamos mažai prisitaikiusios išgyventi gamtoje, tačiau gerai prisitaikiusios prie žmogaus aplinkos. Yra nuomonė, kad mišrūnai yra sveikesni už grynaveislius šunis. Iš tiesų, paveldimos ligos turbūt dažniau pasitaiko naminiams gyvūnams nei laukiniams gyvūnams.
„Vienas iš svarbiausių tikslų yra metodų, skirtų derinti gyvūnų tobulinimo pagal pasirinktus požymius ir jų tinkamumo išlaikyti reikiamu lygiu užduotis, sukūrimas, o ne vienašališka atranka siekiant maksimalaus (kartais perdėto, perdėto) specifinių veislės savybių ugdymo. , kuri yra pavojinga prijaukintų organizmų biologinei gerovei“ – (Lerner, 1958).
Atrankos veiksmingumas, mūsų nuomone, turėtų būti susijęs su sergančių gyvūnų anomalijų diagnozavimu ir nešiotojų, kurių paveldimumas yra trūkumas, bet normalus fenotipas, nustatymas. Sergančių gyvūnų gydymas, siekiant koreguoti jų fenotipus, gali būti vertinamas ne tik kaip įvykis, skirtas gyvūnų estetinei išvaizdai pagerinti (oligodontija), bet ir užkirsti kelią vėžiui (kriptorchizmas), palaikyti biologinį, visavertį aktyvumą (klubo displazija) ir stabilizuoti sveikatą. apskritai. Šiuo atžvilgiu atranka nuo anomalijų būtina bendroje kinologijos ir veterinarijos veikloje.
Galimybė tirti DNR dėl įvairių šunų ligų yra labai naujas dalykas kinologijoje, to žinios gali perspėti veisėjus, į kokias genetines ligas reikėtų atkreipti ypatingą dėmesį renkantis tėvelių poras. Gera genetinė sveikata yra labai svarbi, nes ji lemia biologiškai visavertį šuns gyvenimą. Daktaro Padgetto knygoje „Paveldimų šunų ligų kontrolė“ parodyta, kaip perskaityti genetinę kilmę dėl bet kokių anomalijų. Genetiniai kilmės dokumentai parodys, ar liga yra susijusi su lytimi, ar paveldima per paprastą dominuojantį geną, ar per recesyvinį geną, ar liga yra poligeninė. Kartkartėmis pasitaikys netyčinių genetinių klaidų, nesvarbu, koks rūpestingas veisėjas. Naudojant genetinius kilmės dokumentus kaip įrankį dalytis žiniomis, žalingus genus galima atskiesti tiek, kad jie nebereikštųsi tol, kol bus rastas DNR žymeklis, skirtas patikrinti jų perdavimą. Kadangi atrankos procesas apima populiacijos tobulinimą naujoje kartoje, atsižvelgiama ne į tiesioginių atrankos strategijos elementų (individų ar sukryžmintų individų porų) fenotipines savybes, o į jų palikuonių fenotipines savybes. Būtent dėl šios aplinkybės kyla poreikis apibūdinti požymio paveldėjimą veisimo užduotims atlikti. Kryžminių individų pora nuo kitų panašių individų skiriasi savo kilme ir fenotipinėmis bruožo savybėmis – tiek jie patys, tiek jų giminaičiai. Remiantis šiais duomenimis, jei yra paruoštas paveldėjimo aprašymas, galima gauti numatomas palikuonių savybes, taigi ir kiekvieno veisimo strategijos elemento atrankos verčių įvertinimus. Atliekant bet kokią intervenciją, skirtą bet kokiai genetinei anomalijai, pirmiausia reikia nustatyti „blogojo“ bruožo santykinę svarbą, palyginti su kitais požymiais. Jei nepageidaujamas požymis dažnai paveldimas ir sukelia rimtą žalą šuniui, turėtumėte elgtis kitaip, nei tuo atveju, jei požymis yra retas arba nereikšmingas. Puikus veislės šuo, turintis netinkamą spalvą, išlieka daug vertingesnis tėvas nei vidutinis šuo, turintis tinkamą spalvą.
Ar įmanoma pagimdyti sveiką vaiką, jei mama turi MTHFR geno mutaciją? ir gavo geriausią atsakymą
Atsakymas iš Nightbird[guru]
Motinos MTHFR geno mutacija NĖRA VERDIKTAS.
Beje, įvairiose vietose gali būti mutacijų.
Kai mutantinis MTHFR genas aptinkamas heterozigotinėje būsenoje*, nėra jokios įtikinamos priežasties baimintis. Norint išvengti hiperkoaguliacijos būklių, nėštumo metu rekomenduojama kasdien gerti po 0,4 mg folio rūgšties per dvi dozes, gerai maitintis ir kartą per tris mėnesius (arba pagal indikacijas) tirti hemostazogramą.
Dažniausias fermento defektas, susijęs su vidutiniu HC (homocisteino) koncentracijos padidėjimu, yra geno, koduojančio MTHFR, mutacija. MTHFR katalizuoja folio rūgšties pavertimą aktyvia forma. Iki šiol buvo aprašytos 9 MTHFR geno, esančio 1p36.3 lokuse, mutacijos. Dažniausias iš jų yra C677T pakeitimas (MTHFR baltyme – valino pakeitimas alaninu), pasireiškiantis termolabilumu ir MTHFR fermento aktyvumo sumažėjimu. Pastebėta, kad padidinus folatų kiekį maiste, galima išvengti GC koncentracijos padidėjimo plazmoje.
Homocisteino kiekio padidėjimas kraujo plazmoje tiesiogiai koreliuoja su trombomodulino sintezės slopinimu, AT-III ir endogeninio heparino aktyvumo sumažėjimu, taip pat su tromboksano A2 gamybos aktyvavimu. Ateityje tokie pokyčiai sukelia mikrotrombozę ir mikrocirkuliacijos sutrikimus, o tai savo ruožtu vaidina svarbų vaidmenį spiralinių arterijų patologijoje ir akušerinių komplikacijų, susijusių su gimdos placentos kraujotakos pokyčiais, vystymuisi.
Padidėjusio homocisteino kiekio kraujyje priežastis: MTHFR geno C677T variantas - fermento metilentetrahidrofolato reduktazės geno mutacija.
Citoziną pakeitus timinu 677 padėtyje, fermento funkcinis aktyvumas sumažėja iki 35% vidutinės vertės.
Polimorfizmo duomenys:
*homozigotų atsiradimo dažnis populiacijoje yra 10-12 proc.
*heterozigotų atsiradimo populiacijoje dažnis – 40 proc.
....
T varianto nešiotojai nėštumo metu patiria folio rūgšties trūkumą, o tai lemia vaisiaus nervinio vamzdelio vystymosi defektus.
Rūkymas sustiprina 677T varianto poveikį...
Folio rūgšties vartojimas gali žymiai sumažinti šio polimorfizmo pasekmių riziką.
daugiau informacijos čia --
Apskritai, kas kur bus nuvežtas... Tiksliai pasakyti neįmanoma. Tai priklauso ir nuo tėvo – kas jo genome!!!
Pabandykite užduoti savo klausimą išsamiau čia --
Arba dar geriau čia -
SĖKMĖS!
Daugiau apie studiją
Gilberto sindromas yra paveldima liga, kuriai būdingi geltos epizodai ir padidėjęs nekonjuguoto (laisvo, netiesioginio) bilirubino kiekis kraujo serume. Jo paplitimas siekia apie 5 proc.
Sindromo išsivystymo priežastis – sumažėjęs kepenų fermento uridino difosfato-gliukuroniltransferazės (UDPGT), kurį koduoja genas, aktyvumas. UGT 1A1. Mutacija geno promotoriaus srityje UGT 1A1 būdingas TA pakartojimų skaičiaus padidėjimas (paprastai jų skaičius neviršija 6). Jei homozigotinės ar heterozigotinės būklės yra 7 (ar rečiau 8), UDPGT fermento funkcinis aktyvumas sumažėja – tai būtina sąlyga Gilberto sindromui atsirasti. Homozigotinių mutacijų nešiotojams ligai būdingas didesnis pradinis bilirubino kiekis ir sunkesnės klinikinės apraiškos. Esant heterozigotiniams nešiotojams, vyrauja latentinė ligos forma.
Paprastai raudonųjų kraujo kūnelių irimo metu išsiskiria netiesioginis bilirubinas, kuris turi būti pašalintas iš organizmo. Patekęs į kepenų ląsteles, jis jungiasi su gliukurono rūgštimi, veikiamas fermento uridino difosfato gliukuroniltransferazės (UDPGT). Dėl bilirubino ir gliukurono rūgšties derinio jis tirpsta vandenyje, todėl jis patenka į tulžį ir išsiskiria su šlapimu. Dėl geno mutacijos UGT1 A1 ir dėl nepakankamo UDPGT aktyvumo sutrinka netiesioginio bilirubino konjugacija, todėl padidėja jo koncentracija kraujyje. Padidėjęs bilirubino kiekis kraujyje, savo ruožtu, skatina jo kaupimąsi audiniuose, ypač elastinguose audiniuose (yra kraujagyslių sienelėse, odoje, skleroje) – tuo paaiškinama gelta.
Gilberto sindromo apraiškos gali pasireikšti bet kuriame amžiuje, jas išprovokuoja fizinis aktyvumas, stresinės situacijos, badavimas, virusinės infekcijos, alkoholio vartojimas ir daugybė hepatotoksinį poveikį turinčių vaistų. Liga pasižymi nespecifiniais simptomais: pilvo skausmas, sunkumas dešinėje hipochondrijoje, virškinimo sutrikimai (pykinimas, raugėjimas, vidurių užkietėjimas, viduriavimas), nuovargis, bendras negalavimas, nerimas. Pagrindinis simptomas yra icterinė odos ir gleivinių spalvos pakitimas ir netiesioginio bilirubino kiekio padidėjimas kraujyje. Hiperbilirubinemija (padidėjęs bilirubino kiekis) dažniausiai gali būti ne didesnis kaip 100 mmol/l, vyraujant netiesioginei frakcijai. Likę kepenų tyrimų rodmenys paprastai nesikeičia.
Veikiant saulės šviesai Gilberto sindromu sergantiems pacientams gali padidėti odos pigmentacija.
Kartais liga pasireiškia naujagimio laikotarpiu ir yra vertinama kaip fiziologinė naujagimių gelta.
Galima ir nuolatinė besimptomė eiga, tuomet Gilberto sindromą galima aptikti atsitiktinai aptiktais biocheminio kraujo tyrimo (bilirubino rodiklio) pakitimais.
Laiku diagnozavus Gilberto sindromą, galima atskirti jį nuo kitų kepenų ir kraujo ligų, operatyviai apriboti hepatotoksinį poveikį turinčių vaistų vartojimą, išvengti kepenų krizių, koreguoti paciento gyvenimo būdą, kol visiškai išnyks hiperbilirubinemijos sukeltas diskomfortas.
Greičiausias būdas nustatyti Gilberto sindromą yra tiesioginė DNR diagnozė, kurią sudaro geno TA pasikartojimų skaičiaus nustatymas. UGT1A1.
Veiksniai, provokuojantys Gilberto sindromo paūmėjimą:
- sunkus fizinis aktyvumas,
- mitybos klaidos (konservai, kepti, aštrūs, rūkyti maisto produktai, gazuoti gėrimai),
- badas,
- alkoholis,
- stresinės situacijos, pervargimas,
- insoliacija,
- virusinės infekcijos,
- vaistai, kurių metabolizme dalyvauja UDPGT fermentas (anaboliniai steroidai, gliukokortikoidai, androgenai, etinilestradiolis, rifampicinas, cimetidinas, chloramfenikolis, streptomicinas, chloramfenikolis, natrio salicilatas, ampicilinas, kofeinas, paracetamolis, irinotekanas).
Kada numatytas tyrimas?
- Jei įtariamas Gilberto sindromas.
- Gilberto sindromo ir kitų ligų, pasireiškiančių hiperbilirubinemija, diferencinėje diagnozėje.
- Dėl didelio Gilberto sindromo paplitimo prieš pradedant gydymą vaistais, turinčiais hepatotoksinį poveikį, rekomenduojama atlikti genetinį tyrimą.
- Įvertinti komplikacijų riziką gydymo irinotekanu (vaistu nuo vėžio) metu.
- Dėl lengvos neinfekcinės geltos.
- Kai pacientas serga lėtine gelta, kurią palengvina barbitūratai.
- Jei bilirubino koncentracija padidėja esant kitiems normaliems biocheminiams kraujo parametrams.
- Su šeimos istorija (neinfekcinė gelta, hiperbilirubinemija).
Genetikoje, kaip ir bet kuriame kitame moksle, yra specifinė terminija, skirta paaiškinti pagrindines sąvokas. Dar mokykloje daugelis iš mūsų girdėjo tokius terminus kaip dominavimas, recesyvumas, genas, alelis, homozigotiškumas ir heterozigotiškumas, tačiau iki galo nesupratome, kas už jų slypi. Išsamiau panagrinėkime, kas yra homozigotas, kuo jis skiriasi nuo heterozigotos ir kokį vaidmenį jo formavime atlieka aleliniai genai.
Šiek tiek bendros genetikos
Norėdami atsakyti į klausimą, kas yra homozigotas, prisiminkime Gregoro Mendelio eksperimentus. Kryžmindamas skirtingos spalvos ir formos žirnių augalus, jis padarė išvadą, kad gautas augalas kažkokiu būdu paveldėjo genetinę informaciją iš savo „protėvių“. Nors „geno“ sąvokos dar nebuvo, Mendelis sugebėjo bendrais bruožų paveldėjimo mechanizmą paaiškinti. Devyniolikto amžiaus viduryje Mendelio atrasti dėsniai lėmė tokį teiginį, vėliau pavadintą „lytinių ląstelių grynumo hipoteze“: „Kai susidaro gameta, į ją patenka tik vienas iš dviejų alelinių genų, atsakingų už tam tikrą požymį“. Tai yra, iš kiekvieno iš tėvų gauname tik vieną alelinį geną, atsakingą už tam tikrą požymį – ūgį, plaukų spalvą, akių spalvą, nosies formą, odos atspalvį.
Aleliniai genai gali būti dominuojantys arba recesyviniai. Tai priartina mus prie homozigotos apibrėžimo. Dominuojantys aleliai gali užmaskuoti recesyvą, kad jis nepasireikštų fenotipu. Jei abu genotipo genai yra recesyviniai arba dominuojantys, tai yra homozigotinis organizmas.

Homozigotų tipai
Iš viso to, kas išdėstyta aukščiau, galime atsakyti į klausimą, kas yra homozigotas: tai ląstelė, kurioje už tam tikrą požymį atsakingi aleliniai genai yra vienodi. Aleliniai genai yra homologinėse chromosomose ir, homozigoto atveju, gali būti recesyviniai (aa) arba dominuojantys (AA). Jei vienas alelis yra dominuojantis, o kitas ne, tai yra heterozigotas (Aa). Tuo atveju, kai ląstelės genotipas yra aa, tai yra recesyvinis homozigotas, jei AA yra dominuojantis, nes joje yra alelių, atsakingų už dominuojantį požymį.
Perėjimo ypatybės
Kryžminant du vienodus (recesyvinius arba dominuojančius) homozigotus, susidaro ir homozigotas.
Pavyzdžiui, yra dvi baltos rododendros gėlės su bb genotipais. Juos sukryžminus gausime ir baltą gėlę, kurios genotipas.
Taip pat galite pateikti pavyzdį su akių spalva. Jei abu tėvai turi rudas akis ir yra homozigotiniai šiam požymiui, tai jų genotipas yra AA. Tada visi vaikai turės rudas akis.
Tačiau kryžminant homozigotus ne visada susidaro organizmas, homozigotinis bet kuriam požymiui. Pavyzdžiui, sukryžminus raudonus (DD) ir baltus (dd) gvazdikus, gali atsirasti rožinė arba raudona ir balta gėlė. Rožinis gvazdikas, kaip ir dviejų spalvų gvazdikas, yra nepilno dominavimo pavyzdys. Abiem atvejais gauti augalai bus heterozigotiniai su Dd genotipu.

Homozigotų pavyzdžiai
Gamtoje yra gana daug homozigotų pavyzdžių. Baltos tulpės, gvazdikai, rododendrai yra recesyvinių homozigotų pavyzdžiai.
Žmonėms dėl alelinių genų sąveikos taip pat dažnai susidaro organizmai, kurie yra homozigotiniai pagal kurį nors požymį, nesvarbu, ar tai būtų labai šviesi oda, mėlynos akys, šviesūs plaukai ar daltonizmas.
Dominuojantys homozigotai taip pat dažni, tačiau dėl dominuojančių bruožų gebėjimo užmaskuoti recesyvinius iš karto pasakyti, ar žmogus yra recesyvinio alelio nešiotojas, ar ne, neįmanoma. Dauguma genų, atsakingų už genetines ligas, atsiranda dėl genų mutacijų ir yra recesyviniai, todėl atsiranda tik tuo atveju, jei homologinėse chromosomose nėra normalaus, dominuojančio alelio.
Panašūs straipsniai